

Researchers from Hefei University of Technology in China have recently published an exhaustive review in the Journal of Medicinal Chemistry on emerging roles of small kinase inhibitors in the treatment of diseases. In contrast to well established kinase cancer treatments, their review is focused squarely on nononcologic diseases such as central nervous system disorders, cardiovascular disease, viral infection and autoimmune and inflammatory disease. Since these diseases are not typically the result of cells breaking free of normal kinase controls on proliferation, often through readily detectable escape variants, identifying specific druggable targets can be more challenging.

Protein kinases are now the second-most popular group of drug targets after the G-protein-coupled receptors, primarily due to their success as targeted chemotherapies. As many of the mysteries of once hopelessly interwoven kinase signaling pathways are quickly being unmasked through the miracles of gene editing and experimental mutants, non-oncological diseases are now enjoying an inevitable trickle-down effect from cancer research.
Perhaps the best way to introduce kinase inhibitors is to take a few examples, the big ones, and explore different ways they can be classified. The first kinase inhibitor in the U.S. to be repurposed for nononcologic disease, ruxolitinib (Jakafi), was approved by the FDA in 2011 for treatment of myelofibrosis. It is an inhibitor of Janus kinases (JAK1 & JAK2). Janus kinases are now targets in for treatment of inflammatory disorders included SARS-CoV-2-induced cytokine storm, and inhibitors may even block endocytosis or other steps in the viral life cycle. The suffix ‘-tinib’ in ruxolitinib is a dead giveaway for inhibitors of tyrosine kinases, but then again, most kinase inhibitors act on tyrosine kinases.
The reason for this is that tyrosine kinases are usually plasma membrane proteins that play important roles in signal transduction from outside the cell all the way to the nucleus. Subsequent modifications of gene expression typically affect the metabolism and growth of the cell. Tyrosine kinases came into their element after the respiratory and other critical energy production apparatuses became concentrated within symbiotic organellar membranes like mitochondria, thereby freeing up the external membrane for dedicated sensing and signaling functions.
Ruxolitinib competitively targets the ATP binding site and stabilizes the kinase-active conformation. It is therefore known as a type I inhibitor, as opposed to type II inhibitors, which bind and stabilize the inactive conformation. There are also type i-1/2 inhibitors that bind unique conformations, and type III and IV allosteric inhibitors that bind to other parts of the kinase. Inhibitors can bind their targets covalently or noncovalently. Covalent kinase inhibitors are increasingly sought because of their higher selectivity, sustained target inhibition, improved pharmacodynamics, and better ligand efficiency, but they require a reactive residue near the active site to which they can bind.
Several inhibitors of Bruton’s kinase (BTK), like ibrutinib, are irreversible, covalently binding inhibitors. Imbrutinib targets a cysteine at location 481 of the kinase. BTK is important for the growth, differentiation and activation of myeloid, mast and B cells, and has therefore emerged as an important target for many immune-related diseases. Some successes have already been reported in treating acute respiratory distress in SARS-CoV-2 using Ibrutinib; however, there are toxicity issues to be considered. Imbrutinib achieves a reduction in the produce of mediators of inflammation, including TNFα, IL1, IL-6 and Monocyte chemo-attractant protein-1 by neutrophils and macrophages,
One the most successful small-molecule kinase inhibitors is imatinib (Gleevec), which targets a constitutively activated fusion kinase in CML and ALL. Imatinib has been found to have antiviral activity in some models; however, in a human lung model, it failed to reduce viral replication. Another potential application of imatinib, but not directly related to its kinase inhibitor function, is Alzheimer’s disease. The drug can inhibit gamma-secretase activating protein (GSAP), which selectively increases the production and accumulation of neurotoxic beta-amyloid plaques. Curiously, it appears that imatinib may not even need to cross the blood brain barrier to work, as some studies have indicated production of beta-amyloid initiates in the liver.
Kinase inhibitors have also been developed for serine/threonine kinases, and a few of these have already shown promise for treating neurological diseases and immune disorders. Rho-associated protein kinases (ROCKs), and interleukin-1 receptor-associated kinases (IRAKs) are both ser/thr kinases for which inhibitors are being actively developed. To access the brain, a drug needs to be permeable to the blood-brain barrier and resistant to efflux transporters. ALK (anaplastic lymphoma kinase) inhibitors, like crizotinib, can be made brain penetrant by increasing lipophilicity or using a method called scaffold hopping. ALK is a tyrosine kinase that is highly expressed in the brain and is critical for its proper development.
Another angle in treating disease with kinase inhibitors has been to target unique kinases in organisms that have infected us. To this end, the calcium-dependent protein kinases (Pf CDPKs) of the malarial parasite Plasmodium falciparum, which are the main transducers of calcium-initiated signaling functions, are in the crosshairs. Similar kinases acting in the widespread Toxoplasma gondii parasite have also emerged as key targets. An unusual feature of these parasite-derived CDPKs is a special glycine gatekeeper sitting within its ATP binding pocket that is entirely unprecedented in the human versions of the protein. If inhibitors can be designed that ignore our own CDPKs but block the parasite’s, this would likely make parasite CDPK a safe target.
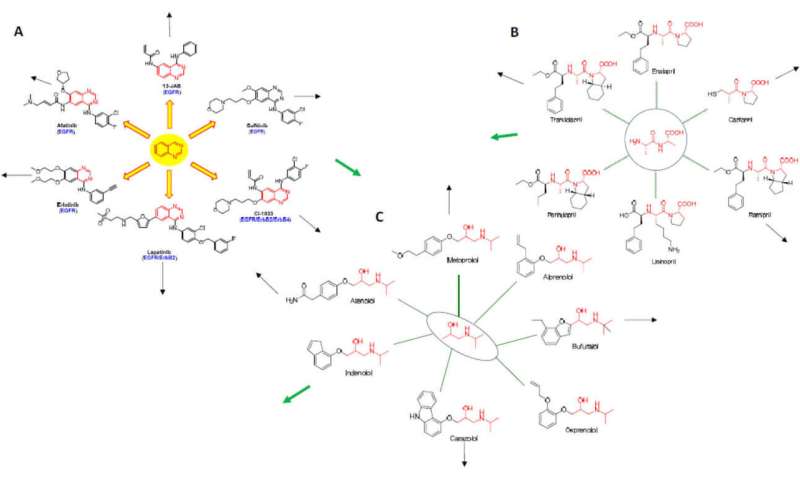
While the rational design of kinase inhibitors is still a bit of an art form, new computational techniques are able to tweak structures to create both highly selective inhibitors and dual and multitarget inhibitors. Graph theory and network analysis is also now used to predict whether a particular drug might be successfully repurposed to other diseases. One approach is based on so-called “Erdos interactomes” wherein drugs (here called nodes), are anchored around their underlying core structural motif. For tyrosine kinase inhibitors, this may be the quinazoline core. First-order or higher-order interactomes are then built up, and their diagrams overlapped with other kinds of drugs to generate new applications.
Although large monoclonal antibodies can be raised to bind and inhibit kinases with high specificity, their high inherent production cost, low stability and intrinsic immunogenicity have reduced them to experimental luxuries for the lab. Instead, small-molecule inhibitors have now become the bread and butter of drug discovery efforts everywhere.
John Hewitt , Medical Xpress